
|
Photophysics and Photochemistry of Transition Metal Compounds |
| Home Research Members Collaborations Publications |



|
||||||||
Ultrafast time-resolved infrared spectroscopy of [Ru(bpy)3]2+ (bpy = 2,2â-bipyridine), [Ru(mbpy)3]2+ (mbpy = 6-methyl-2,2â-bipyridine), and [Ru(mphen)3]2+ (mphen = 2-methyl-1,10â-phenanthroline) in deuterated acetonitrile serves to elucidate the evolution of the system following pulsed excitation into the 1MLCT band at 400 nm. Whereas for [Ru(bpy)3]2+ no intermediate state can be evidenced for the relaxation of the corresponding 3MLCT state back to the ground state, for [Ru(mbpy)3]2+ and [Ru(mphen)3]2+ an intermediate state with a lifetime of about 400 ps is observed. The species associated IR difference spectra of this state are in good agreement with the calculated difference spectra of the lowest energy 3dd state using DFT. The calculated potential energy curves for all the complexes in the triplet manifold along the metal-ligand distance show that for [Ru(bpy)3]2+ the 3dd state is at higher energy than the 3MLCT state and that there is a substantial barrier between the two minima. For [Ru(mbpy)3]2+ and [Ru(mphen)3]2+, the 3dd state is at lower energy than the 3MLCT state. | ||||||||

|
||||||||
The role of ligand-field states for the photophysical properties of d6 systems has been discussed in a large number of publications over the past decades. Since the seminal paper by Houten and Watts, for instance, the quenching of the 3MLCT luminescence in ruthenium(II) polypyridyl complexes is attributed to the presence of the first excited ligand-field state, namely a component of the 3T1(t2g5eg1) state, at similar energies. If this state lies above the 3MLCT state, the luminescence is quenched via thermal population at elevated temperatures only. If it lies well below, then the luminescence is quenched down to cryogenic temperatures. In this contribution we present transient absorption spectra on non-luminescent ruthenium polypyridyl complexes such as [Ru(m-bpy)3]2+, m-bpy = 6-methyl-2,2â-bipyridine, in acetonitrile at room temperature, which reveal an ultra-rapid depopulation of the 3MLCT state but a much slower ground state recovery. We propose that in this and related complexes the methyl groups force longer metal-ligand bond lengths, thus resulting in a lowering of the ligand-field strength such that the 3dd state drops to below the 3MLCT state, and that furthermore the population of this state from the 3MLCT state occurs faster than its decay to the ground state. In addition we demonstrate that in this complex the luminescence can be switched on by external pressure, which we attribute to a destabilisation of the ligand-field state by the pressure due to its larger molecular volume compared to the ground state as well as the 3MLCT state. | ||||||||
|
 |
|||||||
Ultrafast transient absorption spectroscopy serves to identify the 3dd state as intermediate quencher state of the 3MLCT luminescence in the non-luminescent ruthenium complexes [Ru(m-bpy)3]2+ (m-bpy = 6-methyl-2,2â²-bipyridine) and [Ru(tm-bpy)3]2+ (tm-bpy = 4,4â²,6,6â²-tetramethyl-2â²,2â²-bipyridine). For [Ru(m-bpy)3]2+, the population of the 3dd state from the 3MLCT state occurs within 1.6 ps, while the return to the ground state takes 450 ps. For [Ru(tm-bpy)3]2+, the corresponding values are 0.16 and 7.5 ps, respectively. According to DFT calculations, methyl groups added in the 6 and 6â² positions of bipyridine stabilize the 3dd state by â¼4000 cmâ1 each, compared to [Ru(bpy)3]2+. | ||||||||
 |
|
|||||||
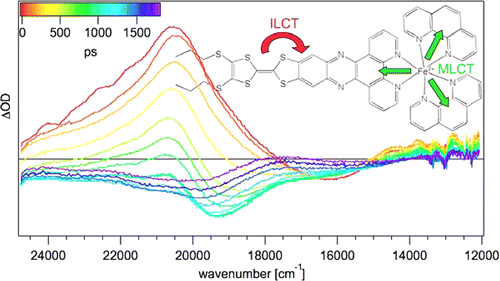
The synthesis and photophysical properties of the complex [Fe(phen)2(TTF-dppz)]2+Â (TTF-dppz = 4â²,5â²-bis-(propylthio)tetrathiafulvenyl[i]dipyrido[3,2-a:2â²,3â²-c]phenazine, phen = 1,10-phenanthroline) are described. In this complex, excitation into the metalâligand charge transfer bands results in the population of a high-spin state of iron(II), with a decay lifetime of approximately 1.5 ns, in dichloromethane, at room temperature. An intraligand charge transfer state can also be obtained and has a lifetime of 38 ps. A mechanism for the different states reached is proposed based on transient absorption spectroscopy. | ||||||||
|
 |
|||||||
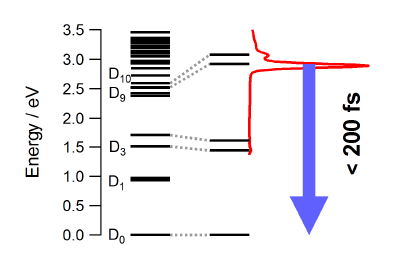
The photophysical properties of the free neutral radical galvinoxyl were studied by a combination of femtosecond time-resolved spectroscopy and quantum chemical calculations. The electronic absorption spectrum is dominated by an intense band at 430 nm that is ascribed to the D9,10âD0 transitions. Upon photoexcitation at 400 nm, the population of the D9,10 states decays within less than 200 fs to the electronic ground state. This ultrafast internal conversion does not involve intramolecular modes with large amplitude motion as the measured dynamics does not show any significant dependence on the environment, but is most probably facilitated by a high density of electronic states of different character. Depending on the solvent, a weak transient band due to the galvinoxylate anion is also observed. This closed-shell species, which is fluorescent although its deactivation is also dominated by non-radiative decay, is generated upon biphotonic ionization of the solvent and electron capture. The ultrashort excited-state lifetime of the galvinoxyl radical precludes photoinduced disproportionation previously claimed to be at the origin of the formation of both anion and cation. | ||||||||
 |
|
|||||||
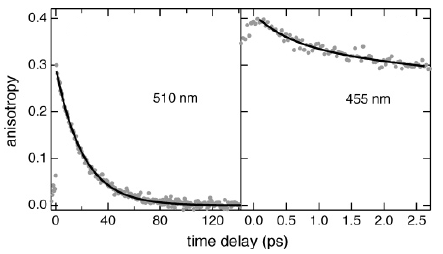
The influence of soluteâsolvent interactions on the vibrational energy relaxation dynamics of perylene and substituted perylenes in the first singlet excited-state upon excitation with moderate (<0.4 eV) excess energy has been investigated by monitoring the early narrowing of their fluorescence spectrum. This narrowing was found to occur on timescales ranging from a few hundreds of femtoseconds to a few picoseconds. Other processes, such as a partial decay of the fluorescence anisotropy and the damping of a low-frequency oscillation due to the propagation of a vibrational wavepacket, were found to take place on a very similar time scale. No significant relationship between the strength of nonspecific soluteâsolvent interactions and the vibrational energy relaxation dynamics of the solutes could be evidenced. On the other hand, in alcohols the spectral narrowing is faster with a solute having H-bonding sites, indicating that this specific interaction tends to favor vibrational energy relaxation. No relationship between the dynamics of spectral narrowing and macroscopic solvent properties, such as the thermal diffusivity, could be found. On the other hand, a correlation between this narrowing dynamics and the number of low-frequency modes of the solvent molecules was evidenced. All these observations cannot be discussed with a model where vibrational energy relaxation occurs via two consecutive and dynamically well-separated steps, namely ultrafast intramolecular vibrational redistribution followed by slower vibrational cooling. On the contrary, the results indicate that both intra- and intermolecular vibrational energy redistribution processes are closely entangled and occur, at least partially, on similar timescales. | ||||||||
|
||||||||
Visible pumpâprobe spectroscopy has been used to identify and characterize short-lived metal-to-metal charge transfer (MMCT) excited states in a group of cyano-bridged mixed-valence complexes of the formula [LCoIIINCMII(CN)5]-, where L is a pentadentate macrocyclic pentaamine (L14) or triamine-dithiaether (L14S) and M is Fe or Ru. Nanosecond pumpâprobe spectroscopy on frozen solutions of [L14CoIIINCFeII(CN)5]- and [L14SCoIIINCFeII(CN)5]- at 11 K enabled the construction of difference transient absorption spectra that featured a rise in absorbance in the region of 350â400 nm consistent with the generation of the ferricyanide chromophore of the photoexcited complex. The MMCT excited state of the Ru analogue [L14CoIIINCRuII(CN)5]- was too short-lived to allow its detection. Femtosecond pumpâprobe spectroscopy on aqueous solutions of [L14CoIIINCFeII(CN)5]- and [L14SCoIIINCFeII(CN)5]- at room temperature enabled the lifetimes of their CoIIâFeIII MMCT excited states to be determined as 0.8 and 1.3 ps, respectively. | ||||||||
Download this list in format RIS
 EndNote
EndNote  BibTex
BibTex  PDF XML
PDF XML Last update Friday December 08 2017